You are browsing environment: HUMAN GUT
CAZyme Information: MGYG000003659_00139
You are here: Home > Sequence: MGYG000003659_00139
Basic Information |
Genomic context |
Full Sequence |
Enzyme annotations |
CAZy signature domains |
CDD domains |
CAZyme hits |
PDB hits |
Swiss-Prot hits |
SignalP and Lipop annotations |
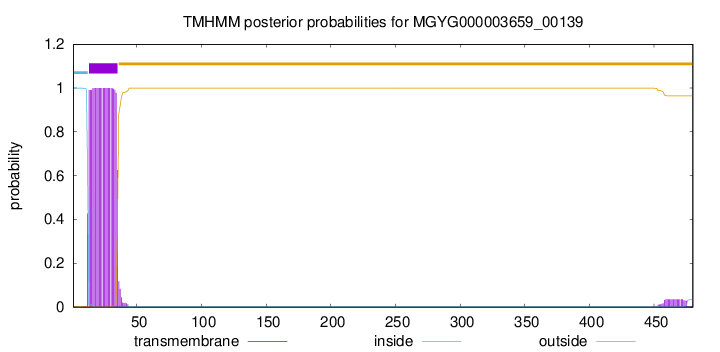
TMHMM annotations
Basic Information help
| Species | CAG-594 sp900771805 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lineage | Bacteria; Firmicutes; Bacilli; RF39; UBA660; CAG-594; CAG-594 sp900771805 | |||||||||||
| CAZyme ID | MGYG000003659_00139 | |||||||||||
| CAZy Family | GT2 | |||||||||||
| CAZyme Description | hypothetical protein | |||||||||||
| CAZyme Property |
|
|||||||||||
| Genome Property |
|
|||||||||||
| Gene Location | Start: 68492; End: 69934 Strand: + | |||||||||||
CAZyme Signature Domains help
| Family | Start | End | Evalue | family coverage |
|---|---|---|---|---|
| GT2 | 236 | 357 | 3.3e-38 | 0.7235294117647059 |
CDD Domains download full data without filtering help
| Cdd ID | Domain | E-Value | qStart | qEnd | sStart | sEnd | Domain Description |
|---|---|---|---|---|---|---|---|
| pfam02397 | Bac_transf | 2.45e-62 | 37 | 182 | 32 | 181 | Bacterial sugar transferase. This Pfam family represents a conserved region from a number of different bacterial sugar transferases, involved in diverse biosynthesis pathways. |
| COG2148 | WcaJ | 6.61e-57 | 38 | 185 | 72 | 224 | Sugar transferase involved in LPS biosynthesis (colanic, teichoic acid) [Cell wall/membrane/envelope biogenesis]. |
| pfam00535 | Glycos_transf_2 | 5.58e-37 | 236 | 356 | 1 | 122 | Glycosyl transferase family 2. Diverse family, transferring sugar from UDP-glucose, UDP-N-acetyl- galactosamine, GDP-mannose or CDP-abequose, to a range of substrates including cellulose, dolichol phosphate and teichoic acids. |
| cd04196 | GT_2_like_d | 2.42e-36 | 236 | 440 | 1 | 214 | Subfamily of Glycosyltransferase Family GT2 of unknown function. GT-2 includes diverse families of glycosyltransferases with a common GT-A type structural fold, which has two tightly associated beta/alpha/beta domains that tend to form a continuous central sheet of at least eight beta-strands. These are enzymes that catalyze the transfer of sugar moieties from activated donor molecules to specific acceptor molecules, forming glycosidic bonds. Glycosyltransferases have been classified into more than 90 distinct sequence based families. |
| cd00761 | Glyco_tranf_GTA_type | 1.67e-33 | 237 | 347 | 1 | 113 | Glycosyltransferase family A (GT-A) includes diverse families of glycosyl transferases with a common GT-A type structural fold. Glycosyltransferases (GTs) are enzymes that synthesize oligosaccharides, polysaccharides, and glycoconjugates by transferring the sugar moiety from an activated nucleotide-sugar donor to an acceptor molecule, which may be a growing oligosaccharide, a lipid, or a protein. Based on the stereochemistry of the donor and acceptor molecules, GTs are classified as either retaining or inverting enzymes. To date, all GT structures adopt one of two possible folds, termed GT-A fold and GT-B fold. This hierarchy includes diverse families of glycosyl transferases with a common GT-A type structural fold, which has two tightly associated beta/alpha/beta domains that tend to form a continuous central sheet of at least eight beta-strands. The majority of the proteins in this superfamily are Glycosyltransferase family 2 (GT-2) proteins. But it also includes families GT-43, GT-6, GT-8, GT13 and GT-7; which are evolutionarily related to GT-2 and share structure similarities. |
CAZyme Hits help
| Hit ID | E-Value | Query Start | Query End | Hit Start | Hit End |
|---|---|---|---|---|---|
| AXH51533.1 | 3.86e-177 | 2 | 480 | 9 | 494 |
| CCO04322.1 | 5.22e-176 | 2 | 477 | 26 | 508 |
| AQW22830.1 | 3.60e-175 | 2 | 480 | 9 | 494 |
| BAK48513.1 | 8.25e-174 | 2 | 477 | 50 | 532 |
| BAK29896.1 | 5.70e-102 | 232 | 480 | 2 | 250 |
PDB Hits download full data without filtering help
| Hit ID | E-Value | Query Start | Query End | Hit Start | Hit End | Description |
|---|---|---|---|---|---|---|
| 5W7L_A | 6.71e-62 | 1 | 190 | 5 | 194 | Structureof Campylobacter concisus PglC I57M/Q175M variant [Campylobacter concisus 13826],5W7L_B Structure of Campylobacter concisus PglC I57M/Q175M variant [Campylobacter concisus 13826] |
| 6H1J_A | 6.41e-17 | 235 | 439 | 22 | 233 | ChainA, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H1J_B Chain B, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H1J_C Chain C, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H21_A Chain A, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H21_B Chain B, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H21_C Chain C, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H2N_A Chain A, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H2N_B Chain B, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H2N_C Chain C, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_A Chain A, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_B Chain B, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_C Chain C, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_D Chain D, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_E Chain E, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_F Chain F, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_G Chain G, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_H Chain H, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_I Chain I, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_O Chain O, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_P Chain P, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_Q Chain Q, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_A Chain A, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_B Chain B, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_C Chain C, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_D Chain D, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_E Chain E, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_F Chain F, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_G Chain G, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_H Chain H, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_I Chain I, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_O Chain O, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_P Chain P, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_Q Chain Q, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_A Chain A, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_B Chain B, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_C Chain C, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_D Chain D, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_E Chain E, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_F Chain F, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_G Chain G, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_H Chain H, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_I Chain I, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_O Chain O, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_P Chain P, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_Q Chain Q, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315] |
| 5HEA_A | 2.18e-16 | 233 | 358 | 5 | 130 | CgTstructure in hexamer [Streptococcus parasanguinis FW213],5HEA_B CgT structure in hexamer [Streptococcus parasanguinis FW213],5HEA_C CgT structure in hexamer [Streptococcus parasanguinis FW213],5HEC_A CgT structure in dimer [Streptococcus parasanguinis FW213],5HEC_B CgT structure in dimer [Streptococcus parasanguinis FW213] |
| 5TZE_C | 2.69e-15 | 236 | 343 | 4 | 114 | Crystalstructure of S. aureus TarS in complex with UDP-GlcNAc [Staphylococcus aureus],5TZE_E Crystal structure of S. aureus TarS in complex with UDP-GlcNAc [Staphylococcus aureus],5TZI_C Crystal structure of S. aureus TarS 1-349 [Staphylococcus aureus],5TZJ_A Crystal structure of S. aureus TarS 1-349 in complex with UDP-GlcNAc [Staphylococcus aureus],5TZJ_C Crystal structure of S. aureus TarS 1-349 in complex with UDP-GlcNAc [Staphylococcus aureus],5TZK_C Crystal structure of S. aureus TarS 1-349 in complex with UDP [Staphylococcus aureus] |
| 1H7L_A | 4.50e-15 | 235 | 334 | 3 | 110 | dTDP-MAGNESIUMCOMPLEX OF SPSA FROM BACILLUS SUBTILIS [Bacillus subtilis],1H7Q_A dTDP-MANGANESE COMPLEX OF SPSA FROM BACILLUS SUBTILIS [Bacillus subtilis],1QG8_A Native (Magnesium-Containing) Spsa From Bacillus Subtilis [Bacillus subtilis],1QGQ_A Udp-manganese Complex Of Spsa From Bacillus Subtilis [Bacillus subtilis],1QGS_A Udp-Magnesium Complex Of Spsa From Bacillus Subtilis [Bacillus subtilis] |
Swiss-Prot Hits download full data without filtering help
| Hit ID | E-Value | Query Start | Query End | Hit Start | Hit End | Description |
|---|---|---|---|---|---|---|
| Q0P9D0 | 6.69e-56 | 1 | 190 | 1 | 189 | Undecaprenyl phosphate N,N'-diacetylbacillosamine 1-phosphate transferase OS=Campylobacter jejuni subsp. jejuni serotype O:2 (strain ATCC 700819 / NCTC 11168) OX=192222 GN=pglC PE=1 SV=1 |
| O32268 | 4.04e-52 | 234 | 452 | 7 | 226 | Putative teichuronic acid biosynthesis glycosyltransferase TuaG OS=Bacillus subtilis (strain 168) OX=224308 GN=tuaG PE=2 SV=1 |
| P71062 | 1.62e-48 | 37 | 186 | 34 | 183 | Uncharacterized sugar transferase EpsL OS=Bacillus subtilis (strain 168) OX=224308 GN=epsL PE=2 SV=1 |
| B5L3F2 | 3.47e-41 | 234 | 445 | 5 | 213 | UDP-Glc:alpha-D-GlcNAc-diphosphoundecaprenol beta-1,3-glucosyltransferase WfgD OS=Escherichia coli OX=562 GN=wfgD PE=1 SV=1 |
| Q077R2 | 4.75e-34 | 233 | 479 | 2 | 243 | UDP-Glc:alpha-D-GlcNAc-diphosphoundecaprenol beta-1,3-glucosyltransferase WfaP OS=Escherichia coli OX=562 GN=wfaP PE=1 SV=1 |
SignalP and Lipop Annotations help
This protein is predicted as OTHER
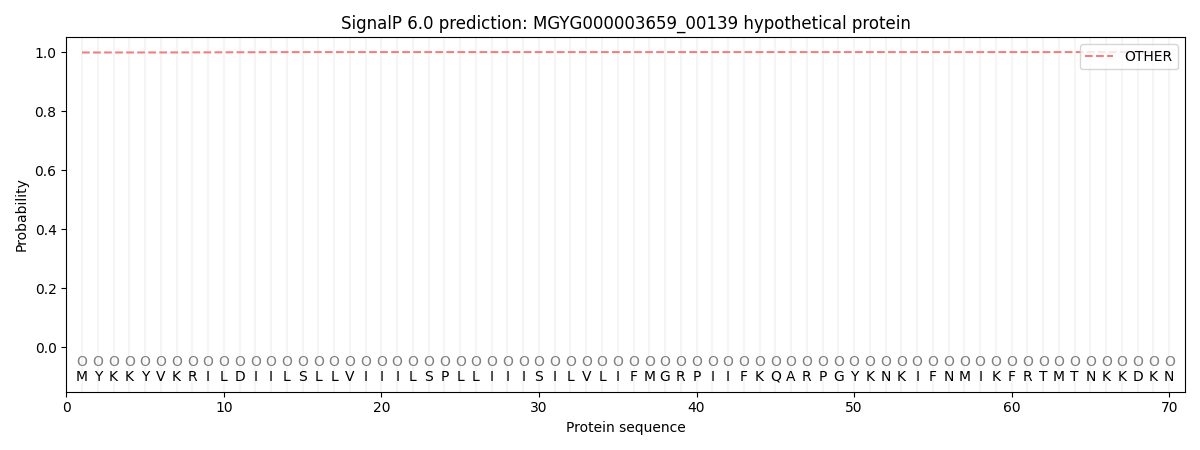
| Other | SP_Sec_SPI | LIPO_Sec_SPII | TAT_Tat_SPI | TATLIP_Sec_SPII | PILIN_Sec_SPIII |
|---|---|---|---|---|---|
| 0.998785 | 0.000695 | 0.000009 | 0.000004 | 0.000002 | 0.000548 |