You are browsing environment: HUMAN GUT
CAZyme Information: MGYG000004905_00404
You are here: Home > Sequence: MGYG000004905_00404
Basic Information |
Genomic context |
Full Sequence |
Enzyme annotations |
CAZy signature domains |
CDD domains |
CAZyme hits |
PDB hits |
Swiss-Prot hits |
SignalP and Lipop annotations |
TMHMM annotations
Basic Information help
| Species | UBA7488 sp002477185 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lineage | Bacteria; Proteobacteria; Alphaproteobacteria; RF32; UBA3637; UBA7488; UBA7488 sp002477185 | |||||||||||
| CAZyme ID | MGYG000004905_00404 | |||||||||||
| CAZy Family | GT2 | |||||||||||
| CAZyme Description | hypothetical protein | |||||||||||
| CAZyme Property |
|
|||||||||||
| Genome Property |
|
|||||||||||
| Gene Location | Start: 381; End: 1286 Strand: - | |||||||||||
CAZyme Signature Domains help
| Family | Start | End | Evalue | family coverage |
|---|---|---|---|---|
| GT2 | 28 | 187 | 1.7e-36 | 0.9764705882352941 |
CDD Domains download full data without filtering help
| Cdd ID | Domain | E-Value | qStart | qEnd | sStart | sEnd | Domain Description |
|---|---|---|---|---|---|---|---|
| pfam00535 | Glycos_transf_2 | 7.75e-39 | 28 | 184 | 1 | 162 | Glycosyl transferase family 2. Diverse family, transferring sugar from UDP-glucose, UDP-N-acetyl- galactosamine, GDP-mannose or CDP-abequose, to a range of substrates including cellulose, dolichol phosphate and teichoic acids. |
| cd00761 | Glyco_tranf_GTA_type | 3.30e-35 | 29 | 216 | 1 | 154 | Glycosyltransferase family A (GT-A) includes diverse families of glycosyl transferases with a common GT-A type structural fold. Glycosyltransferases (GTs) are enzymes that synthesize oligosaccharides, polysaccharides, and glycoconjugates by transferring the sugar moiety from an activated nucleotide-sugar donor to an acceptor molecule, which may be a growing oligosaccharide, a lipid, or a protein. Based on the stereochemistry of the donor and acceptor molecules, GTs are classified as either retaining or inverting enzymes. To date, all GT structures adopt one of two possible folds, termed GT-A fold and GT-B fold. This hierarchy includes diverse families of glycosyl transferases with a common GT-A type structural fold, which has two tightly associated beta/alpha/beta domains that tend to form a continuous central sheet of at least eight beta-strands. The majority of the proteins in this superfamily are Glycosyltransferase family 2 (GT-2) proteins. But it also includes families GT-43, GT-6, GT-8, GT13 and GT-7; which are evolutionarily related to GT-2 and share structure similarities. |
| cd06433 | GT_2_WfgS_like | 1.42e-33 | 28 | 229 | 1 | 196 | WfgS and WfeV are involved in O-antigen biosynthesis. Escherichia coli WfgS and Shigella dysenteriae WfeV are glycosyltransferase 2 family enzymes involved in O-antigen biosynthesis. GT-2 enzymes have GT-A type structural fold, which has two tightly associated beta/alpha/beta domains that tend to form a continuous central sheet of at least eight beta-strands. These are enzymes that catalyze the transfer of sugar moieties from activated donor molecules to specific acceptor molecules, forming glycosidic bonds. Glycosyltransferases have been classified into more than 90 distinct sequence based families. |
| cd04196 | GT_2_like_d | 6.04e-30 | 28 | 232 | 1 | 208 | Subfamily of Glycosyltransferase Family GT2 of unknown function. GT-2 includes diverse families of glycosyltransferases with a common GT-A type structural fold, which has two tightly associated beta/alpha/beta domains that tend to form a continuous central sheet of at least eight beta-strands. These are enzymes that catalyze the transfer of sugar moieties from activated donor molecules to specific acceptor molecules, forming glycosidic bonds. Glycosyltransferases have been classified into more than 90 distinct sequence based families. |
| cd04195 | GT2_AmsE_like | 5.34e-28 | 28 | 227 | 1 | 201 | GT2_AmsE_like is involved in exopolysaccharide amylovora biosynthesis. AmsE is a glycosyltransferase involved in exopolysaccharide amylovora biosynthesis in Erwinia amylovora. Amylovara is one of the three exopolysaccharide produced by E. amylovora. Amylovara-deficient mutants are non-pathogenic. It is a subfamily of Glycosyltransferase Family GT2, which includes diverse families of glycosyltransferases with a common GT-A type structural fold, which has two tightly associated beta/alpha/beta domains that tend to form a continuous central sheet of at least eight beta-strands. These are enzymes that catalyze the transfer of sugar moieties from activated donor molecules to specific acceptor molecules, forming glycosidic bonds. |
CAZyme Hits help
| Hit ID | E-Value | Query Start | Query End | Hit Start | Hit End |
|---|---|---|---|---|---|
| ALN43872.1 | 7.85e-55 | 25 | 229 | 4 | 209 |
| VEI94124.1 | 7.85e-55 | 25 | 229 | 4 | 209 |
| VEJ05099.1 | 7.85e-55 | 25 | 229 | 4 | 209 |
| QIK61386.1 | 1.96e-53 | 23 | 229 | 4 | 206 |
| QIK55991.1 | 3.88e-53 | 23 | 230 | 4 | 207 |
PDB Hits download full data without filtering help
| Hit ID | E-Value | Query Start | Query End | Hit Start | Hit End | Description |
|---|---|---|---|---|---|---|
| 6P61_A | 4.43e-17 | 25 | 134 | 13 | 121 | Structureof a Glycosyltransferase from Leptospira borgpetersenii serovar Hardjo-bovis (strain JB197) [Leptospira borgpetersenii serovar Hardjo-bovis str. JB197],6P61_B Structure of a Glycosyltransferase from Leptospira borgpetersenii serovar Hardjo-bovis (strain JB197) [Leptospira borgpetersenii serovar Hardjo-bovis str. JB197],6P61_C Structure of a Glycosyltransferase from Leptospira borgpetersenii serovar Hardjo-bovis (strain JB197) [Leptospira borgpetersenii serovar Hardjo-bovis str. JB197],6P61_D Structure of a Glycosyltransferase from Leptospira borgpetersenii serovar Hardjo-bovis (strain JB197) [Leptospira borgpetersenii serovar Hardjo-bovis str. JB197] |
| 5HEA_A | 2.24e-16 | 26 | 116 | 6 | 95 | CgTstructure in hexamer [Streptococcus parasanguinis FW213],5HEA_B CgT structure in hexamer [Streptococcus parasanguinis FW213],5HEA_C CgT structure in hexamer [Streptococcus parasanguinis FW213],5HEC_A CgT structure in dimer [Streptococcus parasanguinis FW213],5HEC_B CgT structure in dimer [Streptococcus parasanguinis FW213] |
| 1H7L_A | 1.20e-15 | 25 | 141 | 1 | 124 | dTDP-MAGNESIUMCOMPLEX OF SPSA FROM BACILLUS SUBTILIS [Bacillus subtilis],1H7Q_A dTDP-MANGANESE COMPLEX OF SPSA FROM BACILLUS SUBTILIS [Bacillus subtilis],1QG8_A Native (Magnesium-Containing) Spsa From Bacillus Subtilis [Bacillus subtilis],1QGQ_A Udp-manganese Complex Of Spsa From Bacillus Subtilis [Bacillus subtilis],1QGS_A Udp-Magnesium Complex Of Spsa From Bacillus Subtilis [Bacillus subtilis] |
| 6H1J_A | 3.52e-14 | 27 | 205 | 22 | 202 | ChainA, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H1J_B Chain B, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H1J_C Chain C, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H21_A Chain A, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H21_B Chain B, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H21_C Chain C, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H2N_A Chain A, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H2N_B Chain B, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H2N_C Chain C, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_A Chain A, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_B Chain B, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_C Chain C, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_D Chain D, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_E Chain E, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_F Chain F, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_G Chain G, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_H Chain H, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_I Chain I, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_O Chain O, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_P Chain P, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4F_Q Chain Q, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_A Chain A, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_B Chain B, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_C Chain C, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_D Chain D, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_E Chain E, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_F Chain F, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_G Chain G, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_H Chain H, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_I Chain I, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_O Chain O, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_P Chain P, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6H4M_Q Chain Q, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_A Chain A, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_B Chain B, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_C Chain C, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_D Chain D, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_E Chain E, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_F Chain F, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_G Chain G, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_H Chain H, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_I Chain I, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_O Chain O, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_P Chain P, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315],6HNQ_Q Chain Q, Probable ss-1,3-N-acetylglucosaminyltransferase [Staphylococcus aureus subsp. aureus N315] |
| 3L7I_A | 3.44e-10 | 28 | 205 | 5 | 183 | Structureof the Wall Teichoic Acid Polymerase TagF [Staphylococcus epidermidis RP62A],3L7I_B Structure of the Wall Teichoic Acid Polymerase TagF [Staphylococcus epidermidis RP62A],3L7I_C Structure of the Wall Teichoic Acid Polymerase TagF [Staphylococcus epidermidis RP62A],3L7I_D Structure of the Wall Teichoic Acid Polymerase TagF [Staphylococcus epidermidis RP62A] |
Swiss-Prot Hits download full data without filtering help
| Hit ID | E-Value | Query Start | Query End | Hit Start | Hit End | Description |
|---|---|---|---|---|---|---|
| Q57287 | 5.71e-33 | 25 | 245 | 5 | 233 | Uncharacterized glycosyltransferase HI_1578 OS=Haemophilus influenzae (strain ATCC 51907 / DSM 11121 / KW20 / Rd) OX=71421 GN=HI_1578 PE=3 SV=1 |
| Q58457 | 1.18e-30 | 24 | 227 | 7 | 212 | Uncharacterized glycosyltransferase MJ1057 OS=Methanocaldococcus jannaschii (strain ATCC 43067 / DSM 2661 / JAL-1 / JCM 10045 / NBRC 100440) OX=243232 GN=MJ1057 PE=3 SV=2 |
| P71054 | 3.85e-23 | 25 | 141 | 5 | 122 | Putative glycosyltransferase EpsE OS=Bacillus subtilis (strain 168) OX=224308 GN=epsE PE=2 SV=2 |
| Q1RIM7 | 7.09e-20 | 27 | 236 | 293 | 502 | Uncharacterized glycosyltransferase RBE_0706 OS=Rickettsia bellii (strain RML369-C) OX=336407 GN=RBE_0706 PE=3 SV=1 |
| Q4UM29 | 1.79e-18 | 27 | 236 | 295 | 504 | Uncharacterized glycosyltransferase RF_0543 OS=Rickettsia felis (strain ATCC VR-1525 / URRWXCal2) OX=315456 GN=RF_0543 PE=3 SV=1 |
SignalP and Lipop Annotations help
This protein is predicted as OTHER
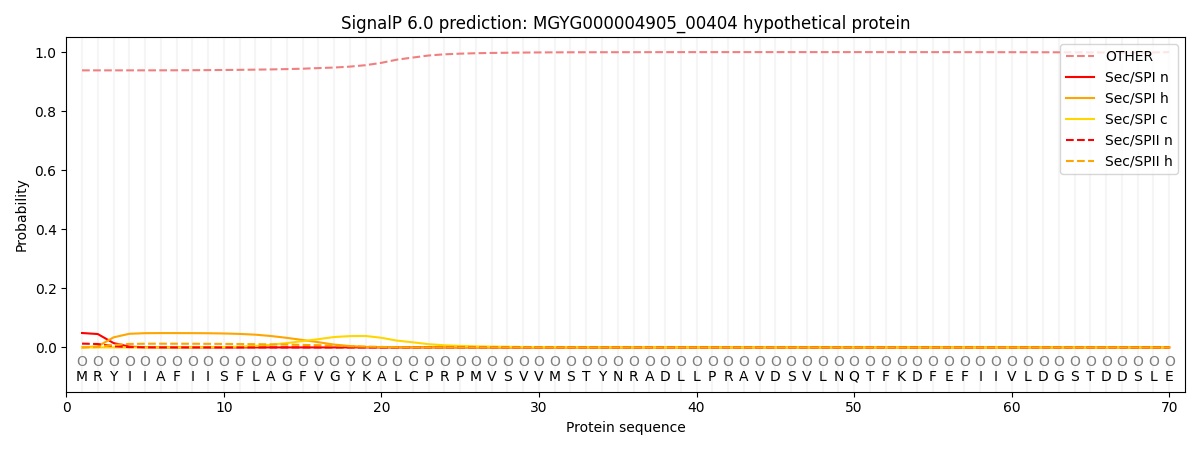
| Other | SP_Sec_SPI | LIPO_Sec_SPII | TAT_Tat_SPI | TATLIP_Sec_SPII | PILIN_Sec_SPIII |
|---|---|---|---|---|---|
| 0.941432 | 0.044527 | 0.013156 | 0.000220 | 0.000122 | 0.000554 |